Lecture
14
Hidden
Markov Model
December
4
Lecturer:
Larry Ruzzo
Notes:
Zizhen Yao
Recall from last
time, in weight matrix method, we assume independent probability of letters
occurring at each position, and fixed sequence length. To capture the dependence
among positions, we need more sophisticated model. Markov and Hidden Markov
Models are the ones widely used.
CpG island prediction.
Let’s look at an interesting biology problem
first. As result of methylation, in general, CpG dinucleotides are rarer in the
genome than would be expected from the independent probability of C and G. In
some region of genome, such as around the promoters or ‘start’ regions of many
gene, we see many more CpG dinucleotides than elsewhere, and in fact more C and
G nucleotides in general. The question we are interested in are:
1. Given a short stretch of genome sequence, how would we decide if it comes from a CpG island or not?
2. Given a long piece of sequence, how would we find the CpG islands in it, if there are any?
Markov Model
In Markov model, the probability of a state depends only on previous state. The model is characterized by the paramters
ast=P( xi=t
|xi-1=s)
A
Markov model (first order) generating DNA sequence look like this:
B:
beginning state. e: ending
state
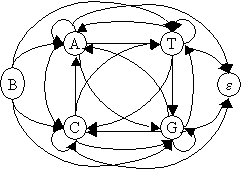
People also use higher order Markov model, in which a state depends only on last few previous states.
To solve CpG island problem, we can create two Markov models: one for CpG island, and one for non-CpG island, each has different transition probabilities on edges. And we compute which one of them is more likely to produce the given DNA sequence.
Given the Markov model, the probability to generate an
output string ![]() is
is
![]()
![]()
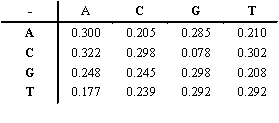 The transition probabilities
are specified in the table below.
The transition probabilities
are specified in the table below.

The log-odds
ratio is given as follows:
![]()
Declare some
threshold for this ratio. If the S(x)
is above the threshold, then it is CpG island, otherwise not.
However, this
approach is unable to answer the second the question: how to pick up the
islands from a long sequence. We could just compute the statistics for all
lengths of , say, 500 bases, but this won’t give us sharp boundaries. In this
case, we need Hidden Markov Model.
Hidden Markov Model:
We can combine the above two models (+, -) , such
every state in + model (except the beginning and end state) has an edge to
every state in – model ( except the beginning and end) and vice versa. Now there are two states corresponding to each
nucleotide symbol. A+, C+, G+, T+ emit
A, C, G, T respectively in CpG island regions, and A-, C-, G-, T -
correspondingly in non-island regions. There is on longer a one-to-one
correspondence between the states and the symbols. We cannot tell what state
the model was by looking at the symbol at that position. That is why it is
referred to as Hidden Markov Model.
 Following is another
interesting application of Hidden Markov Model:
Following is another
interesting application of Hidden Markov Model:
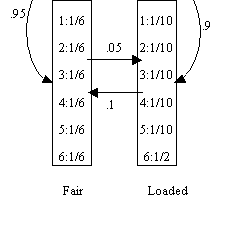
Occasionally
dishonest Casino dice: a Casino uses loaded dice occasionally, while most of
the time using fair dice. The transition graph of fair and loaded state is
given below, with emission probability in the box.
We are interested
to know, given a sequence of throw data, can we tell when a fair dice is used,
and when a loaded dice is used? In general, a related question is “given the
observed data, what is the most likely path through the model?”
Viterbi
Viterbi algorithm
provides solution to the above question. It computes a sequence of state
![]()
given the transition probability and the emission probability ![]() .
.
![]()
The
algorithm uses dynamic programming to compute the probability of the most probable path ending in
state k with observation i, for each k and i. Let
it be ![]() .
.
The
recursive relation is:
![]()
The
details of the algorithm will be discussed next time. We can predict the state
given an observation i, by checking the state at the corresponding
position in the most probable path.